RNA-Seq DE ワークフローまとめ
注意: この演習の読み込みに少し時間がかかる場合があります。
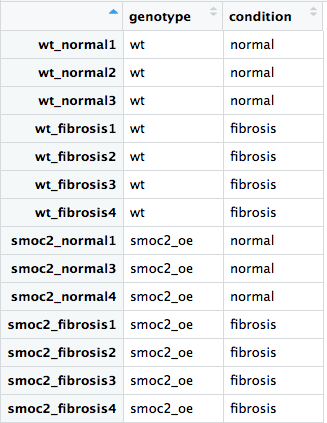
ワイルドタイプと smoc2 過剰発現のサンプルをすべて含むデータセットを使って、DESeq2 のワークフローを実行しましょう。DESeq2 と dplyr ライブラリの読み込み、メタデータファイル all_metadata、および生カウントファイル all_rawcounts の読み込みはあらかじめ済んでいます。
この演習はコースの一部です
RNA-Seq with Bioconductor in R
演習の手順
rownames()、colnames()、all()、および%in%演算子を使って、all_rawcountsとall_metadataのサンプルの順序が一致していることを確認します。genotypeの影響を制御しながらconditionの効果を検定する適切なデザインを指定して、DESeq2 オブジェクトを作成します。genotypeとconditionをそれぞれ制御しつつ、genotype:conditionを検定する適切なデザインを指定して、DESeq2 オブジェクトを作成します。
実践的なインタラクティブ演習
このサンプルコードを完成させて、この演習に挑戦してみましょう。
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)