Résumé du workflow d’analyse DE pour l’ARN-Seq
REMARQUE : Le chargement de cet exercice peut prendre un peu plus de temps.
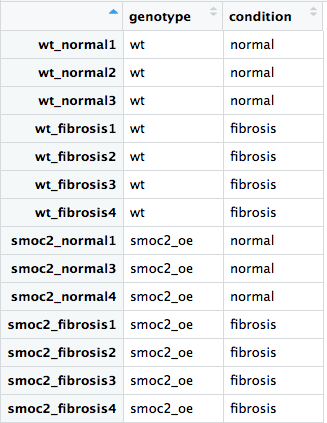
Parcourons le workflow DESeq2 en utilisant l’ensemble de données complet, qui inclut à la fois les échantillons de type sauvage et ceux en sur-expression de smoc2. Nous avons chargé pour vous les bibliothèques DESeq2 et dplyr, ainsi que le fichier de métadonnées all_metadata et le fichier de comptes bruts all_rawcounts.
Cet exercice fait partie du cours
<cours>RNA-Seq avec Bioconductor en R</cours>Instructions de l’exercice
- Vérifiez que les échantillons sont dans le même ordre dans
all_rawcountsetall_metadataen utilisantrownames(),colnames(),all()et l’opérateur%in%. - Créez l’objet DESeq2 avec le design approprié, en testant l’effet de
conditiontout en contrôlantgenotype. - Créez l’objet DESeq2 avec le design approprié, en contrôlant séparément
genotypeetcondition, mais en testantgenotype:condition.
Exercice interactif pratique
Essayez cet exercice en complétant ce code d’exemple.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)