Samenvatting RNA-Seq DE-workflow
LET OP: Het kan iets langer duren om deze oefening te laden.
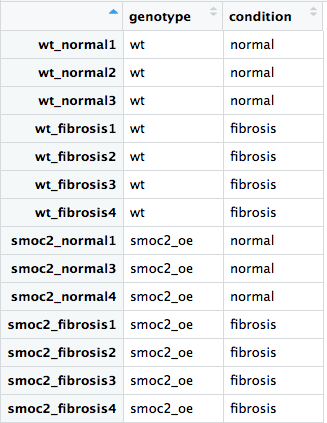
Laten we de DESeq2-workflow doorlopen met de volledige gegevensset, met zowel wildtype- als smoc2-overexpressiemonsters. We hebben de bibliotheken DESeq2 en dplyr alvast geladen en het metadata-bestand all_metadata en het ruwe telbestand all_rawcounts voor je ingelezen.
Deze oefening maakt deel uit van de cursus
RNA-Seq met Bioconductor in R
Oefeninstructies
- Controleer of de monsters in dezelfde volgorde staan in zowel
all_rawcountsalsall_metadatametrownames(),colnames(),all()en de operator%in%. - Maak het DESeq2-object met het juiste design: test het effect van
conditionterwijl je corrigeert voorgenotype. - Maak het DESeq2-object met het juiste design waarbij je afzonderlijk corrigeert voor
genotypeencondition, maar test voorgenotype:condition.
Interactieve oefening met praktijkervaring
Probeer deze oefening door deze voorbeeldcode aan te vullen.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)