Zusammenfassung des RNA-Seq-DE-Workflows
HINWEIS: Das Laden dieser Übung kann etwas länger dauern.
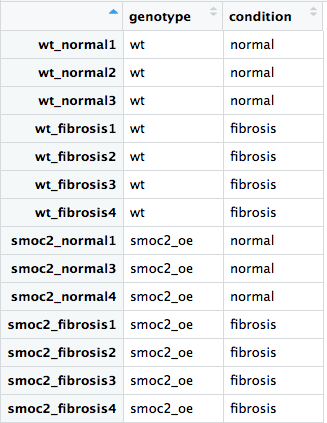
lass uns den DESeq2-Workflow mit dem vollständigen Datensatz durchlaufen, der sowohl Wildtyp- als auch smoc2-Überexpressionsproben enthält. Wir haben die Bibliotheken DESeq2 und dplyr geladen sowie die Metadatendatei all_metadata und die Rohzähldatei all_rawcounts für dich eingelesen.
Diese Übung ist Teil des Kurses
<Kurs>RNA-Seq mit Bioconductor in R</Kurs>Übungsanweisungen
- Prüfe, ob die Proben in
all_rawcountsundall_metadatain derselben Reihenfolge vorliegen, und verwende dazurownames(),colnames(),all()und den Operator%in%. - Erzeuge das DESeq2-Objekt mit dem passenden Design: teste den Effekt von
conditionund kontrolliere dabei fürgenotype. - Erzeuge das DESeq2-Objekt mit dem passenden Design, indem du für
genotypeundconditionjeweils kontrollierst, aber fürgenotype:conditiontestest.
Interaktive praktische Übung
Versuche dich an dieser Übung, indem du diesen Beispielcode vervollständigst.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)