RNA-Seq DE iş akışı özeti
NOT: Bu egzersizin yüklenmesi biraz daha uzun sürebilir.
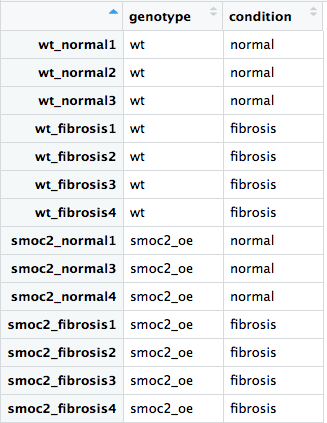
Vahşi tip ve smoc2 aşırı ifade örneklerinin her ikisini de içeren tam veri kümesini kullanarak DESeq2 iş akışını birlikte çalıştıralım. Senin için DESeq2 ve dplyr kütüphanelerini yükledik ve all_metadata adlı üstveri dosyasını ve all_rawcounts adlı ham sayım dosyasını okuduk.
Bu egzersiz, kursun bir parçasıdır
R ile Bioconductor kullanarak RNA-Seq
Egzersiz talimatları
rownames(),colnames(),all()ve%in%işleçlerini kullanarak örneklerin hemall_rawcountshem deall_metadataiçinde aynı sırada olup olmadığını kontrol et.- Uygun tasarımı kullanarak DESeq2 nesnesini oluştur:
genotypeiçin kontrol ederkenconditionetkisini test et. - Uygun tasarımı kullanarak DESeq2 nesnesini oluştur:
genotypeveconditiondeğişkenlerini ayrı ayrı kontrol et, ancakgenotype:conditioniçin test yap.
Uygulamalı etkileşimli egzersiz
Bu egzersizi bu örnek kodu tamamlayarak deneyin.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)