Resumo do fluxo de trabalho de DE em RNA-Seq
OBSERVAÇÃO: Carregar este exercício pode demorar um pouco mais.
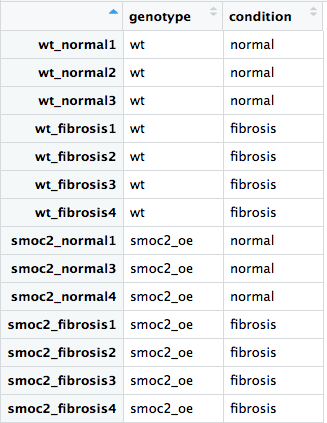
Vamos percorrer o fluxo de trabalho do DESeq2 usando o conjunto de dados completo, com amostras de tipo selvagem (wildtype) e com superexpressão de smoc2. Já carregamos as bibliotecas DESeq2 e dplyr e lemos o arquivo de metadados, all_metadata, e o arquivo de contagens brutas, all_rawcounts, para você.
Este exercicio faz parte do curso
RNA-Seq com Bioconductor em R
Instruções do exercicio
- Verifique se as amostras estão na mesma ordem em
all_rawcountseall_metadatausandorownames(),colnames(),all()e o operador%in%. - Crie o objeto do DESeq2 usando o design apropriado, testando o efeito de
conditionenquanto controla porgenotype. - Crie o objeto do DESeq2 usando o design apropriado, controlando individualmente por
genotypeecondition, mas teste paragenotype:condition.
exercicio interativo prático
Tente este exercicio completando este código de exemplo.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)