Ringkasan alur kerja DE RNA-Seq
CATATAN: Memuat latihan ini mungkin memerlukan waktu sedikit lebih lama.
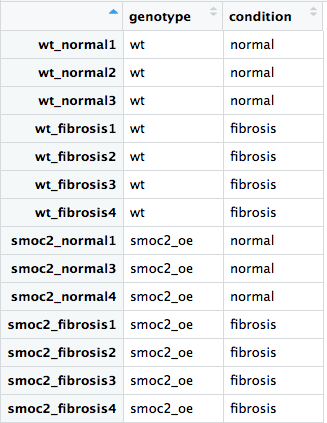
Mari jalankan alur kerja DESeq2 menggunakan himpunan data lengkap yang mencakup sampel wildtype dan overekspresi smoc2. Kami telah memuat pustaka DESeq2 dan dplyr serta membaca berkas metadata, all_metadata, dan berkas hitungan mentah, all_rawcounts, untuk Anda.
Latihan ini merupakan bagian dari kursus
RNA-Seq dengan Bioconductor di R
Instruksi latihan
- Periksa bahwa urutan sampel sama di
all_rawcountsdanall_metadatamenggunakanrownames(),colnames(),all(), dan operator%in%. - Buat objek DESeq2 menggunakan desain yang sesuai, menguji efek
conditionsambil mengontrolgenotype. - Buat objek DESeq2 menggunakan desain yang sesuai, mengontrol
genotypedanconditionsecara individual, tetapi uji untukgenotype:condition.
Latihan interaktif langsung praktik
Cobalah latihan ini dengan melengkapi kode contoh ini.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)