Resumen del flujo de trabajo de DE en RNA-Seq
NOTA: Cargar este ejercicio puede tardar un poco más.
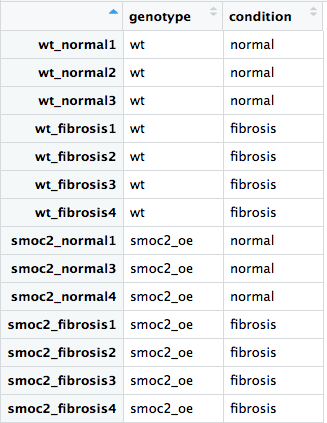
Recorramos el flujo de trabajo de DESeq2 usando el conjunto de datos completo, que incluye tanto muestras wildtype como con sobreexpresión de smoc2. Hemos cargado por ti las librerías DESeq2 y dplyr, y hemos leído el archivo de metadatos all_metadata y el archivo de cuentas sin procesar all_rawcounts.
Este ejercicio forma parte del curso
RNA-Seq con Bioconductor en R
Instrucciones del ejercicio
- Comprueba que las muestras están en el mismo orden en
all_rawcountsyall_metadatausandorownames(),colnames(),all()y el operador%in%. - Crea el objeto de DESeq2 usando el diseño adecuado, evaluando el efecto de
conditionmientras controlas porgenotype. - Crea el objeto de DESeq2 usando el diseño adecuado, controlando por
genotypeyconditionindividualmente, pero realizando la prueba paragenotype:condition.
ejercicio interactivo práctico
Prueba este ejercicio completando este código de ejemplo.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)