Creating the DE object
NOTE: It may take a bit longer to load this exercise.
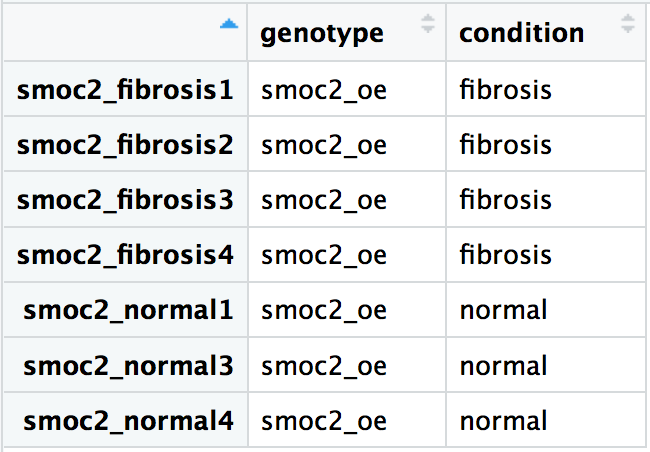
Using our smoc2 overexpression samples, create the DESeq2 object such that the design formula specifies the comparison of the expression differences between the fibrosis and normal samples. The metadata for the experiment is displayed below. We have the data read in with the samples in the same order for the smoc2 raw counts, reordered_smoc2_rawcounts, and the metadata, smoc2_metadata.
This exercise is part of the course
RNA-Seq with Bioconductor in R
Exercise instructions
Create a DESeq2 object called
dds_smoc2using theDESeqDataSetFromMatrix()function by specifying the arguments:countData,colData, anddesign.Run the
DESeq()function to estimate the size factors, calculate the dispersions, and perform the model fitting and testing.
Hands-on interactive exercise
Have a go at this exercise by completing this sample code.
# Create DESeq2 object
dds_smoc2 <- ___(___ = ___,
___ = ___,
___ = ~ condition)
# Run the DESeq2 analysis
___ <- ___(___)