RNA-Seq DE workflow summary
NOTE: It may take a bit longer to load this exercise.
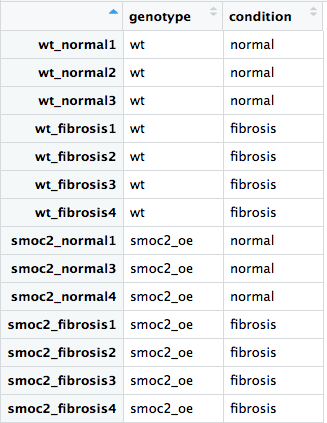
Let's run through the DESeq2 workflow using the full dataset with both wildtype and smoc2 overexpression samples included. We have loaded the DESeq2 and dplyr libraries and read in the metadata file, all_metadata and the raw counts file, all_rawcounts for you.
This exercise is part of the course
RNA-Seq with Bioconductor in R
Exercise instructions
- Check that the samples are in the same order in both
all_rawcountsandall_metadatausing therownames(),colnames(),all(), and%in%operator. - Create the DESeq2 object using the appropriate design, testing for the effect of
conditionwhile controlling forgenotype. - Create the DESeq2 object using the appropriate design, controlling for
genotypeandconditionindividually, but test forgenotype:condition.
Hands-on interactive exercise
Have a go at this exercise by completing this sample code.
# Check that all of the samples are in the same order in the metadata and count data
all(___(___) %in% ___(___))
# DESeq object to test for the effect of fibrosis regardless of genotype
dds_all <- DESeqDataSetFromMatrix(countData = ___,
colData = ___,
design = ___)
# DESeq object to test for the effect of genotype on the effect of fibrosis
dds_complex <- DESeqDataSetFromMatrix(countData = ___,
___,
___)